Syndrome
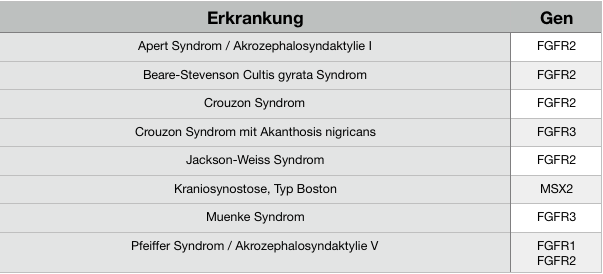
Unterschiedliche klinische Merkmale der FGFR-bedingten Kraniosynostosen
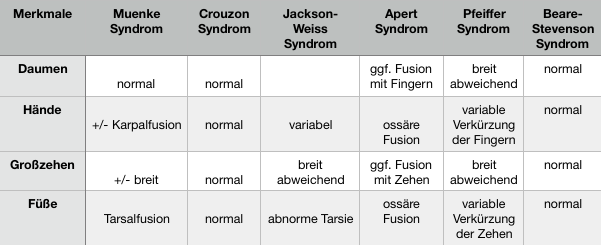
Apert-Syndrom
Das Apert-Syndrom gehört zu den kraniofazialen Fehlbildungen was zu vielfältigen körperlichen Dysplasien führt. In Deutschland leben etwa 400 Menschen mit diesem Syndrom.
Mögliche Symptome sind:
Kopfbereich
- Verwachsen von Schädelknochen mit der Gefahr eines Drucks auf das Gehirn und
Bildung eines Hydrozephalus (Kraniosynostose)
- Fehlbildung des Oberkiefers
- offene Gaumenspalte
- eingeschränktes Hörvermögen
- Sehbehinderung
- Epilepsie
- eingeschränkte Atmung
Hals
- häufig Segmentationsstörungen der HWS
Extremitäten
- zusammengewachsene Finger und Zehen
- versteifte oder fehlende Mittelgelenke
Knochenbau
- Eingeschränkte Bewegung der Gelenke
- Verkrümmung der Wirbelsäule (Skoliose)
Psychologisch
- Probleme bei sozialer und emotionaler Entwicklung (anders sein)
Die genetischen Informationen befinden sich im Zellkern einer Körperzelle auf den Chromosomen. Diese bestehen aus leiterförmigen DNA-Strängen, den eigentlichen Trägern der Erbinformation. Deshalb wird eine genetische Untersuchung auch DNA-Analyse genannt. Gene sind einzelne Abschnitte eines DNA Strangs.
Beare-Stevenson Cultis gyrata Syndrom
Das Beare-Stevenson Syndrom ist extrem selten, bis jetzt sind nur einzelne Fälle in der Literatur beschrieben. Das Auftreten erfolgte immer durch Spontanmutation, eine familiäre Häufung wurde bisher nicht beschrieben.
Mögliche Symptome sind:
- Weit auseinander stehende Augen
- Gaumenspalte
- Vorzeitige Fusion der Schädelknochen (Kraniosynostose)
- Hervortretende Augen
Crouzon Syndrom
Der autosomal-dominant vererbte Morbus Crouzon wird in einem von 25.000 Geburten beschrieben. Die geistige Entwicklung der Kinder läuft meist normal.
Mögliche Symptome sind:
- Schädeldeformitäten, Kraniosynostose
- Vorspringende Schädelnähte
- Exophthalmus (hervorstehende Augäpfel)
- Hypertelorismzs (vergrößerter Augenabstand), spominente Stirn
- Strabismus divergens (Schielen)
- maxiläre Retrognathie (Oberkieferhypoplasie)
- Vorstehende Unterlippe
Jackson-Weiss Syndrom
Das Jackson-Weiss Syndrom ist eine sehr seltene vererbbare Erkrankung mit einer Kombination von Kraniosynostose, Hypoplasie des Mittelgesichtes und Synostosen der Füße. Die Häufigkeit wird mit unter 1 zu 1.000.000 angegeben, die Vererbung erfolgt autosomal-dominant.
Mögliche Symptome sind:
- Kraniosynostose in unterschiedlicher Ausprägung
- Gesichtsdysmorphie
- Synostose am Rück- oder Mittelfuß (tarsal oder metatarsal)
- Verbreitete Großzehe
Kraniosynostose, Typ Boston
Dieses Kraniosynostose-Syndrom wurde in einer Familie bei 19 Mitgliedern aus drei Generationen beschrieben. Die Krankheit wird autosomal-dominant mit hoher Penetranz vererbt. Die Intelligenz erleidet keinerlei Einschränkung.
Mögliche Symptome sind:
- Balkonstirn
- Kleeblattschädel
- schwere Kopfschmerzen
- Sehstörungen
- verkürzte erste Mittelfußknochen
Muenke Syndrom
Das Muenke Syndrom, ist eine vererbliche Form einer Kraniosynostose, die Koronarnaht betreffend mit zusätzlicher Veränderung der Hand- und Fußwurzelknochen.
Die Häufigkeit ist nicht bekannt, eine Koronarnahtsynostose tritt bei etwa 1 von 15.000 Neugeborenen auf. Für das Muenke Syndrom bedarf es des Nachweisen der Genmutation.
Mögliche Symptome sind:
- Durch den Vorzeitigen Verschluss der Koronarnaht ergibt sich eine veränderte Kopf-
und Gesichtsform. - Fusion von Hand- und Fußwurzelknochen
Pfeiffer Syndrom
Das Pfeiffer Syndrom ist eine seltene, autosomal-dominant vererbte Krankheit die nicht mit dem Kardiokranialen Syndrom Typ Pfeife zu verwechseln ist.
Betroffen ist etwa 1 von 100.000 Personen
Mögliche Symptome sind:
- Kurzer Schädel
- Flacher Hinterkopf
- Ausgeprägte Stirn
- Großer Augenabstand
- Unterentwickeltes Mittelgesicht
- Flache Nasenwurzel
- Tiefsitzende Ohren
- Breite, nach außen gerichtete Endglieder von Daumen und Großzehen
- Teilweise sind Zeige- und Mittelfinger und der 2. bis 4. Zeh zusammengewachsen
- Mittelglieder der Finger sind verkürzt
- Dreiecksform der Daumen- und Großzehengrundglieder
Es werden drei Typen des Syndrom unterschieden
Quellenangabe: genetik-dresden.de, Wikipedia, orpha.net